Contributing to Cool-Seq-Tool#
Bug reports and feature requests#
Bugs and new feature requests can be submitted to the Cool-Seq-Tool issue tracker on GitHub. See this StackOverflow post for tips on how to craft a helpful bug report.
Development Setup#
Clone the repository:
git clone https://github.com/genomicmedlab/cool-seq-tool
cd cool-seq-tool
Create a virtual environment and install all dependency groups:
python3 -m venv venv
source venv/bin/activate
python3 -m pip install -e ".[dev,tests,docs]"
We use pre-commit to run conformance tests before commits. This provides checks for:
Code format and style
Added large files
AWS credentials
Private keys
Before your first commit, run:
pre-commit install
Style#
Code style is managed by Ruff, and should be checked via pre-commit hook before commits. Final QC is applied with GitHub Actions to every pull request.
Tests#
Tests are executed with pytest:
pytest
Docstring code snippets are also runnable as doctests:
pytest --doctest-modules src/
Documentation#
The documentation is built with Sphinx, which is included in the docs dependency group. Navigate to the docs/ subdirectory and use make to build the HTML version:
# in an existing virtualenv environment
python3 -m pip install -e '.[docs]'
cd docs
make html
See the Sphinx documentation for more information.
transcript_mappings.tsv#
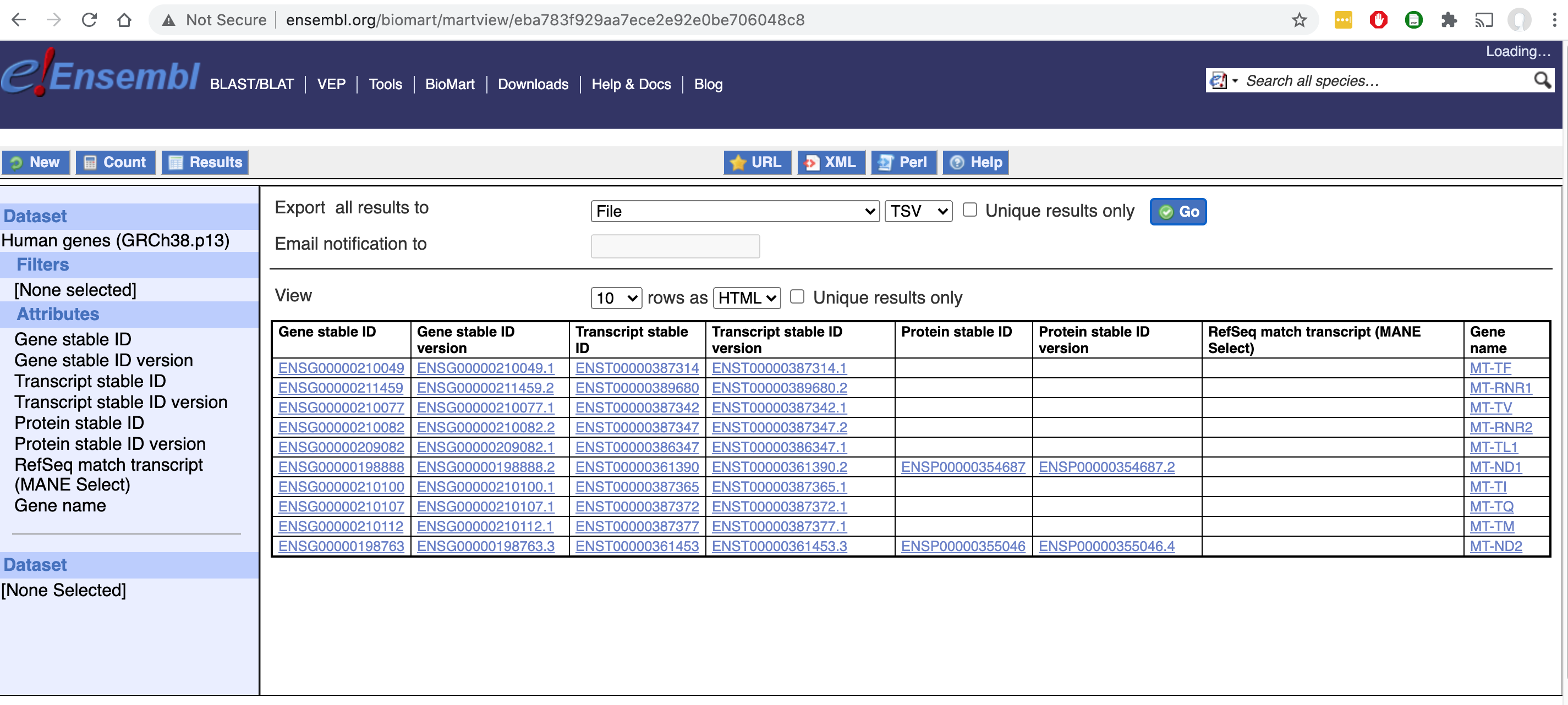
Cool-Seq-Tool uses a static copy of transcript mapping data acquired from Ensembl BioMart. To regenerate this file from the BioMart, select the Human Genes (GRCh38.p13) dataset and choose the following attributes:
Gene stable ID
Gene stable ID version
Transcript stable ID
Transcript stable ID version
Protein stable ID
Protein stable ID version
RefSeq match transcript (MANE Select)
Gene name
Alternatively, use wags-tails:
>>> from wags_tails import EnsemblTranscriptMappingData
>>> data_fetcher = EnsemblTranscriptMappingData()
>>> data, version = data_fetcher.get_latest()